Case of the Month ...
Case History
The patient is an 18 year-old white male who presented with a 4.5 cm right anterior cervical mass, which he first discovered 2 months ago. The mass was firm, mobile and non-tender. He complained of a sore throat and right ear pain, but denied having night sweats, fever or weight loss. The mass did not resolve with antibiotics. Fine needle aspiration of the mass was performed.
Diagnosis & Discussion
click on image for larger version
Image Figs:
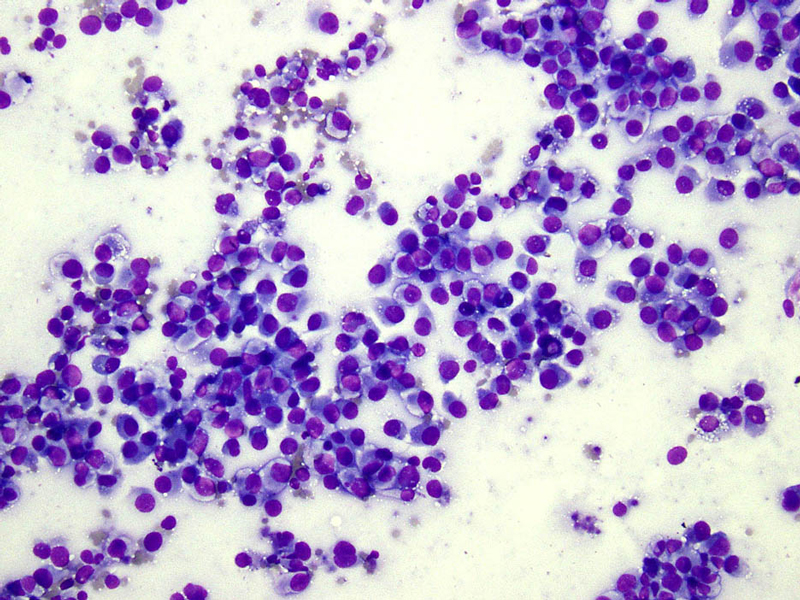
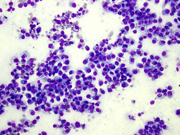
- Figure 1 FNA smear Diff-quik stain 20x
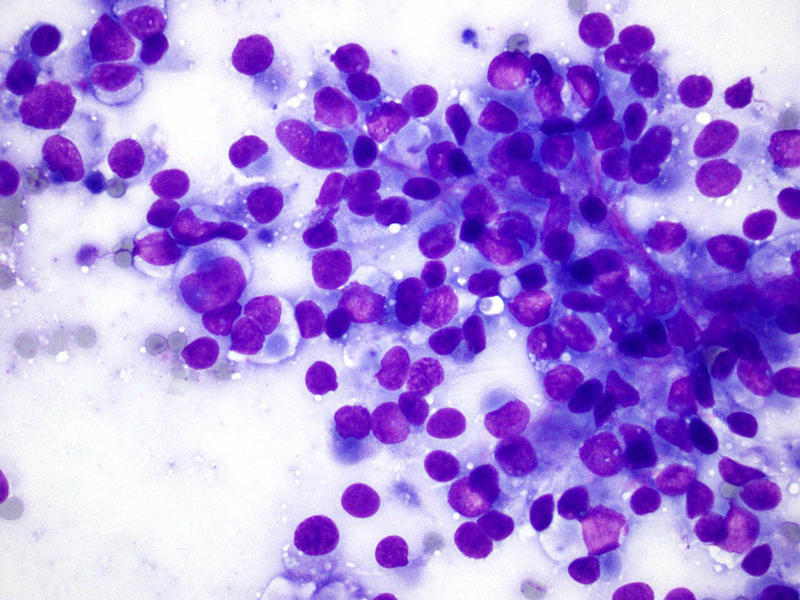
- Figure 2 FNA smear Diff-quik stain 40x
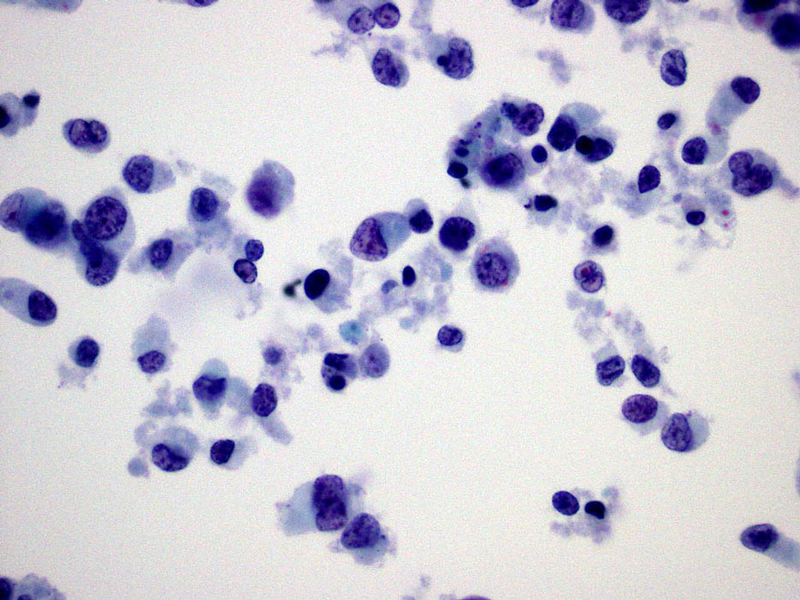
- Figure 3 Thin prep Pap stain 60x
- Figure 4 Thin prep Pap stain - 40x
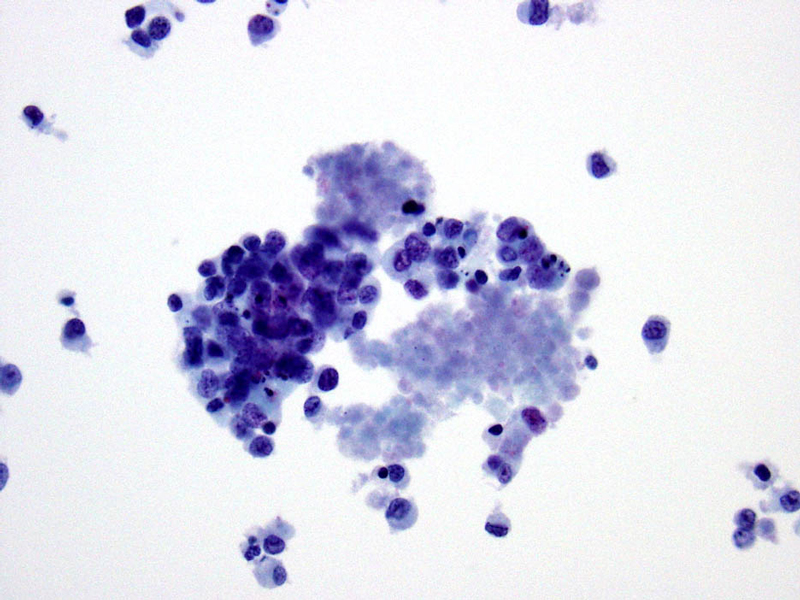
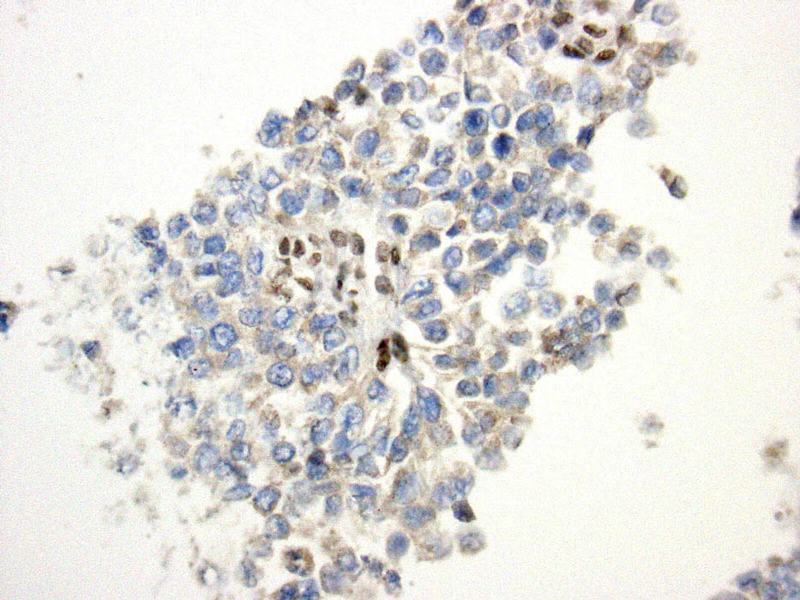
- Figure 5 Section of cell block Immunostain for BAF47 40x
Questions:
- What is the diagnosis?
- Medullary thyroid carcinoma
- Rhabdomyosarcoma
- Proximal-type epithelioid sarcoma
- Synovial sarcoma
- Which of the following tumors retains nuclear expression of BAF47 by immunohistochemistry?
- Malignant rhabdoid tumor
- Renal medullary carcinoma
- Epithelioid sarcoma
- Melanoma
- Which tumor suppressor gene is commonly inactivated in epithelioid sarcomas?
- SMAD4
- SMARCB1/INI1
- MEN1
- RB1
Discussion:
Epithelioid sarcoma (ES) is a malignant soft tissue tumor of uncertain histogenesis with an epithelioid cytologic appearance. The classical type of ES usually occurs in the distal extremities of young adults. It is seen more often in men than women (approximately 2:1), and the median age is 26 years.(1) Proximal-type ES was first described by Guillou et al as an aggressive variant which manifests in more deep-seated, proximal body sites and in slightly older patients.(2) Proximal-type ES typically involves the pelvis, perineum or genital tract, but it can also occur in the head, neck or trunk.
The cytologic appearance of classical type ES shows a mix of epithelioid and spindle cells which are present singly or in loose clusters. Necrosis is seen in the background, and a granulomatous process is often in the differential diagnosis. In proximal-type ES, the aspirate contains predominantly epithelioid cells, many of which have rhabdoid features. The rhabdoid cells have abundant, eosinophilic cytoplasm with paranuclear hyaline-like inclusions which displace and indent the nucleus. The nuclei are round to oval with vesicular chromatin and inconspicuous to prominent nucleoli. Necrosis may also be present.
ES is usually an aggressive tumor with high rates of local recurrence. The rate of recurrence varies from 38% to 77%, while the rate of metastasis has been reported to be between 44% and 47%. (3, 4, 5) ES has a five year survival rate of approximately 66-70%, and a ten year survival rate of 50% . (4,6) Hasegawa et al found a higher rate of metastasis and worse outcome in proximal-type epithelioid sarcoma with 75% of patients developing metastases and 65% dying from their disease during a mean follow-up period of 4 years and 5 months.(7) Other adverse prognostic factors include male gender, non-distal extremity tumor, tumor size greater than or equal to 5cm, increased tumor depth, high mitotic index, hemorrhage, necrosis, vascular invasion and inadequate initial excision. (8)
The standard treatment is complete surgical resection with negative margins. Chemotherapy and/or radiotherapy may be given depending on size of tumor, depth and margin status.
The differential diagnosis for this tumor based on morphologic appearance and tumor site included medullary thyroid carcinoma, rhabdomyosarcoma, and epithelioid sarcoma, as well as carcinoma, melanoma and other mesenchymal neoplasms which may exhibit rhabdoid features. Immunocytochemical stains were helpful in narrowing down the differential diagnosis as the tumor cells were positive for EMA, focally positive for a pancytokeratin (AE1/AE3) and Cam 5.2, and negative for calcitonin, synaptophysin, chromogranin, TTF-1, desmin, CD34, S100, CD45 and BAF47 (INI1). Loss of INI1 nuclear expression has been shown in extrarenal rhabdoid tumor, epithelioid sarcoma, renal medullary carcinoma, epithelioid malignant peripheral nerve sheath tumor and myoepithelial carcinoma of the soft tissues and has rarely been seen in other types of malignancies. (9,10) The loss of INI1 expression by immunocytochemistry reflects a deletion or mutation in the SMARCB1/INI1 gene on chromosome 22q11.2. The SMARCB1/INI1 gene encodes for a subunit of the SWI/SNF chromatin remodeling complex and is thought to act as a tumor suppressor gene. (11)
The tumor in this case showed loss of BAF47 (INI1) and was positive for cytokeratins, which limited the differential diagnosis to epithelioid sarcoma, extrarenal rhabdoid tumor and renal medullary carcinoma. The patient had no renal mass. Proximal-type ES and extrarenal rhabdoid tumor have a similar histologic appearance, but extrarenal rhabdoid tumors occur almost exclusively in infants and small children. Furthermore, the excisional biopsy of our case showed an epithelioid tumor with a nodular appearance and areas of necrosis, both of which are characteristic features of ES.
Answers
- c
- d
- b
REFERENCES
Guillou L and Kaneko Y. Tumors of uncertain differentiation. Epithelioid sarcoma. In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization classification of tumours. Pathology and genetics of tumours of soft tissue and bone. Lyon, France: IARC Press, 2002. p 205-207.
Guillou L, Wadden C, Coindre JM, et al. "Proximal-type" epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol 1997;21:130-146.
Halling AC, Wollan PC, Pritchard DJ, et al. Epithelioid sarcoma: a clinicopathologic review of 55 cases. Mayo Clin Proc 1996;71:636-642.
Ross HM, Lewis JJ, Woodruff JM, et al. Epithelioid sarcoma: clinical behavior and prognostic factors of survival. Ann Surg Oncol 1997;4:491-495.
Chase DR and Enzinger FM. Epithelioid sarcoma: diagnosis, prognostic indicators, and treatment. Am J Surg Pathol 1985;9:241-263.
Bos GD, Pritchard DJ, Reiman HM, et al. Epithelioid sarcoma. An analysis of fifty-one cases. J Bone Joint Surg Am 1988;70:862-870.
Hasegawa T, Matsuno Y, Shimoda T, et al. Proximal-type epithelioid sarcoma: a clinicopathologic study of 20 cases. Mod Pathol 2001;14:655-663.
Weiss SW, Goldblum JR. Enzinger and Weiss's Soft Tissue Tumors. St. Louis, Missouri: Mosby, Inc; 2001.
Sigauke E, Rakheja D, Maddox DL, et al. Absence of expression of SMARCB1/INI1 in malignant rhabdoid tumors of the central nervous system, kidneys and soft tissue: an immunohistochemical study with implications for diagnosis. Mod Pathol 2006;19:717-725.
Hornick JL, Cin PD, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 2009;33:542-550.
Roberts CW, Orkin SH. The SWI/SNF complex - chromatin and cancer. Nat Rev Cancer 2004;4:133-142.
Contritbuted by:
Suzanne M. Brandt MD
Andre L. Moreira MD, PhD
Memorial Sloan Kettering Cancer Center
1275 York Avenue, New York, NY 10065